- Predict pKa, powder X-ray diffraction and crystal morphology
- Calculate Young’s and shear moduli to aid in the optimization of tableting conditions
- Understand solubility in non-aqueous solvents
- Simulate spectroscopy including VCD, NMR (solution and solid-state), IR, Raman, and UV-Vis

Pharmaceutical Formulations & Delivery
Optimize your pharmaceutical at the molecular level
A smart, strategic drug formulation can efficiently advance your drug development projects and inform downstream processes. Advances in molecular modeling and machine learning are enabling atomistic-level insights to improve drug formulations and the ability to evaluate large numbers of candidate materials and formulations prior to experiments.
Schrödinger offers a range of computational solutions for advancing pharmaceutical formulation, from crystalline or amorphous form characterization to selection of materials and excipients for processing, formulation, and delivery.

Intuitive computational workflows designed by experts in formulation chemistry

Easy-to-use system builders for complex formulations of large molecular systems
Powerful workflows for molecular simulation, machine learning, and data analysis
Dedicated customer support and extensive training resources
Key Capabilities
Optimize drug process development and manufacturing with predictive characterization
Understand drug stability and reactivity
- Predict glass transition temperature and water uptake in amorphous materials, including amorphous solid dispersions
- Evaluate drug stability with respect to various degradation channels
- Calculate bond dissociation energy to evaluate chemical stability
- Design molecular catalysts with automated solutions
Predict solubility of drug candidates
- Accurately predict solubility of amorphous and crystalline forms to encourage the discovery of a soluble active pharmaceutical ingredient (API) and to delineate the potential solubility boost from non-crystalline forms using FEP+
- Identify instances where pure drug solubility can exceed the expected solubility due to the formation of small drug aggregates
Characterize and optimize drug formulations and delivery
- Gain insight into the complex requirements and behaviors of lipid-based and polymer-based formulations, including amorphous solid dispersions
- Evaluate the impact of different polymers or polymer residues on the release solubilization and aggregation of the API
- Predict key properties such as hygroscopicity, viscosity and miscibility of ingredients, molecular interactions in solution, and drug release profiles
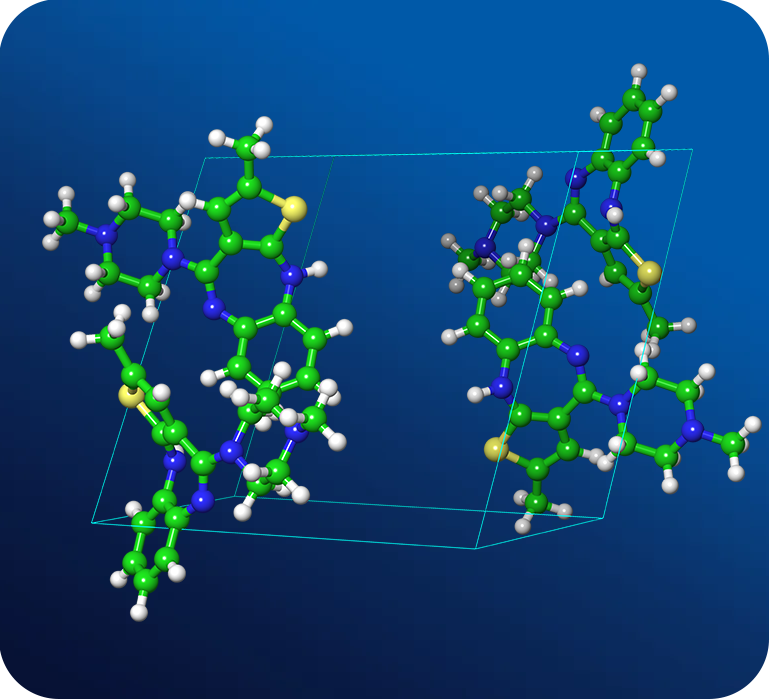
Crystal Structure Prediction Services
De-risk your solid form selection process by identifying the most stable polymorph at room temperature
Overcome the risks associated with disappearing polymorphs in late stage drug development. For a given active pharmaceutical ingredient (API), we will leverage our proprietary crystal structure prediction (CSP) platform to identify the most stable crystal polymorph at room temperature. Starting from a 2D structure of the API, we deliver to you the thermodynamic stability ranking of crystal polymorphs.
Case Studies & Webinars
Discover how Schrödinger technology is being used to solve real-world research challenges.
Advancing the design and optimization of drug formulations with combined computational and experimental approaches
Computationally-Guided Drug Formulation Webinar Series
Characterizing lipid nanoparticle self-assembly and structure using coarse-grained simulations
Advanced Machine Learning and Molecular Simulations for Formulation Design
Tackling drug solubility: AbbVie and Schrödinger collaborate to advance accurate prediction methods
Beyond the lab: Unleashing the potential of in silico modeling in drug product formulation
Advancing the design and optimization of drug formulations with coarse-grained molecular simulations
Computer-aided Formulation Development for Small-molecule Drugs
Featured course

Learn in silico drug formulation methods with our hands-on online certification course
Level-up your skills by enrolling in our online course, Molecular Modeling for Materials Science: Pharmaceutical Formulations.
Learn MoreDocumentation & Tutorials
Get answers to common questions and learn best practices for using Schrödinger’s software.

Key Products
Learn more about the key computational technologies available to progress your research projects.
Virtual Cluster
Secure, scalable environment for running simulations on the cloud
Desmond
High-performance molecular dynamics (MD) engine providing high scalability, throughput, and scientific accuracy
MS CG
Efficient coarse-grained (CG) molecular dynamics (MD) simulations for large systems over long time scales
Jaguar
Quantum mechanics solution for rapid and accurate prediction of molecular structures and properties
Crystal Structure Prediction Services
De-risk your solid form selection process by identifying the most stable polymorph at room temperature
Publications
Browse the list of peer-reviewed publications using Schrödinger technology in related application areas.
Calculating Apparent pKa Values of Ionizable Lipids in Lipid Nanoparticles
Hamilton N et al. ChemRxiv, Mar 2024.
Coarse-Grained Simulation of mRNA-Loaded Lipid Nanoparticle Self-Assembly
Grzetic D et al. ChemRxiv, Feb 2024.
A Robust Crystal Structure Prediction Method to Support Small Molecule Drug Development with Large Scale Validation and Prospective Studies
Zhou D et al. ChemRxiv. January 2024.
Free Energy Perturbation Approach for Accurate Crystalline Aqueous Solubility Predictions
Hong R et al. J. Med. Chem. 2023, 66, 23, 15883–15893.
Drug Aggregation of Sparingly-Soluble Ionizable Drugs: Molecular Dynamics Simulations of Papaverine and Prostaglandin F2α
Skrdla P et al. Mol. Pharmaceutics. 2023, 20, 10, 5135–5147.
Toward a Combined Molecular Dynamics and Quantum Mechanical Approach to Understanding Solvent Effects on Chemical Processes in the Pharmaceutical Industry: The Case of a Lewis Acid-Mediated SNAr Reaction
Tanoury G et al. Org. Process Res. Dev. 2023, 27, 4, 742–754.
A Computational-Based Approach to Fabricate Ceritinib Co-Amorphous System Using a Novel Co-former Rutin for Bioavailability Enhancement
Yarlagadda D et al. European Journal of Pharmaceuticals and Biopharmaceuticals. 2023, 190, 220-230.
Molecular-Level Examination of Amorphous Solid Dispersion Dissolution
Afzal M et al. Mol. Pharmaceutics 2021, 18, 11, 3999–4014.
Dissolution Behavior of Weakly Basic Pharmaceuticals from Amorphous Dispersions Stabilized by a Poly(dimethylaminoethyl Methacrylate) Copolymer
Derek S. Frank, Prateek Prasad, Luca Iuzzolino, and Luke Schenck, Mol. Pharmaceutics 2022, 19, 9, 3304–3313.
Software and services to meet your organizational needs
Software Platform
Deploy digital drug discovery workflows using a comprehensive and user-friendly platform for molecular modeling, design, and collaboration.
Modeling Services
Leverage Schrödinger’s computational expertise and technology at scale to advance your projects through key stages in the drug discovery process.
Support & Training
Access expert support, educational materials, and training resources designed for both novice and experienced users.