
- Publication
- Aug 16, 2022
Induced-Fit Docking Enables Accurate Free Energy Perturbation Calculations in Homology Models
Tianchuan Xu, et al. Journal of Chemical Theory and Computation, 2022, 18(9), 5710-5724
- Publication
- Jul 21, 2022
Discovery of Potent and Orally Bioavailable Pyridine N-Oxide-Based Factor XIa Inhibitors through Exploiting Nonclassical Interactions
Xu, et al. Journal of Medicinal Chemistry, 2022, 65(15), 10419-10440
- Publication
- May 23, 2022
Sequence-based Viscosity Prediction for Rapid Antibody Engineering
Estes, et al. biomolecules, 2024, 14(6), 617
- Publication
- May 23, 2022
Exploring the Activity Profile of TbrPDEB1 and hPDE4 Inhibitors Using Free Energy Perturbation
Lorena Zara, et al. ACS Medicinal Chemistry Letters, 2022, 13(6), 904-910
- Publication
- May 18, 2022
Separating clinical antibodies from repertoire antibodies, a path to in silico developability assessment
Negron, et al. Taylor & Francis, 2022, 14(1), 1-9
- Publication
- May 13, 2022
Structure-based assessment and druggability classification of protein-protein interaction sites
Lara Alzyoud, et al. Scientific Reports, 2023, 12, 7975
- Publication
- Apr 28, 2022
Discovery of a Novel Class of d-Amino Acid Oxidase Inhibitors Using the Schr’dinger Computational Platform
Haifeng Tang, et al. Journal of Medicinal Chemistry, 2022, 65(9), 6775-6802
- Publication
- Apr 13, 2022
AutoDesigner, a De Novo Design Algorithm for Rapidly Exploring Large Chemical Space for Lead Optimization: Application to the Design and Synthesis of D-Amino Acid Oxidase Inhibitors
Pieter H. Bos, et al. JCIM, 2022, 62(8), 1905-1915
- Publication
- Mar 15, 2022
Transferable Neural Network Potential Energy Surfaces for Closed-Shell Organic Molecules: Extension to Ions
Leif D. Jacobson, et al. JCTC, 2022, 18(4), 2354-2366
- Publication
- Mar 11, 2022
A Descriptor Set for Quantitative Structure-Property Relationship Prediction in Biologics
Kannan Sankar, et al. Mol Inform, 2022
- Publication
- Feb 24, 2022
Plasticity in ligand recognition at somatostatin receptors
Michael J. Robertson, et al. Nature Structural & Molecular Biology, 2022, 29, 210-217
Webinars
 Webinar
Life Science
Webinar
Life Science
- Apr 23, 2025
Computational strategies for discovering and optimizing RNA- and DNA-targeting molecules
In this webinar, we will showcase how Schrödinger’s advanced computational solutions are enabling accurate and efficient targeting of RNA and DNA systems by small molecules.
 Webinar
Life Science
Webinar
Life Science
- Apr 16, 2025
Schrödinger デジタル創薬セミナー: Into the Clinic~計算化学がもたらす創薬プロセスの変貌~第16回
Accelerating protein degrader discovery: Computational strategies for degrader design and optimization
 Webinar
Life Science
Materials Science
Webinar
Life Science
Materials Science
- Apr 8, 2025
Accelerating pharmaceutical formulations using machine learning approaches
In this webinar, we will demonstrate how Schrödinger’s integrated ML- and physics-based approaches are transforming pharmaceutical formulation design.
Documentation
- Documentation
Online Help and Documentation
An online resource of information and instruction on how to use Schrödinger software including user manuals, panel descriptions, installation guides, reference sheets, tutorials, and more.
 Documentation
Life Science
Documentation
Life Science
Tutorials
- Tutorial
BACE1 Inhibitor Design Using Free Energy Perturbation
Prepare, run, and analyze a free energy perturbation (FEP) simulation for a series of BACE1 inhibitors using FEP+.
- Tutorial
Ligand Binding Pose Prediction for FEP+ using Core-Constrained Docking
Generate starting poses for FEP simulations for a series of BACE1 inhibitors using core constrained docking
- Tutorial
Introduction to MD Simulations with Desmond
Prepare, run, and perform simple analysis on an all-atom MD simulation with Desmond.
Training Videos
 Video
Life Science
Video
Life Science
Getting Going with Maestro BioLuminate
A free video series introducing the basics of using Maestro Bioluminate.
 Video
Life Science
Video
Life Science
 Video
Life Science
Video
Life Science
A Sneak Peek into Renumbering Residues and the Project Table in BioLuminate
The sixth video in the Getting Going with Maestro BioLuminate Video Series: renaming chains and residues, the Project Table.
Publications
- Publication
- Mar 5, 2025
A robust crystal structure prediction method to support small molecule drug development with large scale validation and blind study
Zhou, et al. Nature Communications, 2025, 16, 2210
- Publication
- Feb 28, 2025
Exploiting solvent exposed salt-bridge interactions for the discovery of potent inhibitors of SOS1 using free-energy perturbation simulations
Leffler, et al. ACS Medicinal Chemistry Letters, 2025
- Publication
- Feb 20, 2025
Predicting Resistance to Small Molecule Kinase Inhibitors
Nagarajan, et al. Journal of Chemical Information and Modeling, 2025, Preprint
Case Studies
 Case Study
Life Science
Case Study
Life Science
 Case Study
Life Science
Case Study
Life Science
 Case Study
Life Science
Case Study
Life Science
White Papers
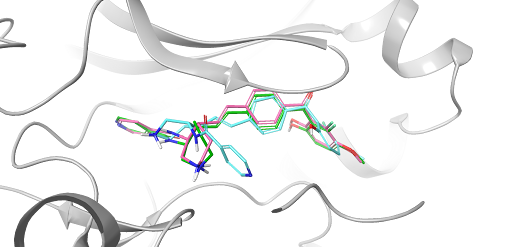 White Paper
Life Science
White Paper
Life Science
- Oct 29, 2024
20 Years of Glide: A Legacy of Docking Innovation and the Next Frontier with Glide WS
Glide has long set the gold standard for commercial molecular docking software due to its robust performance in both binding mode prediction and empirical scoring tasks, ease of use, and tight integration with Schrödinger’s Maestro interface and molecular discovery workflows.
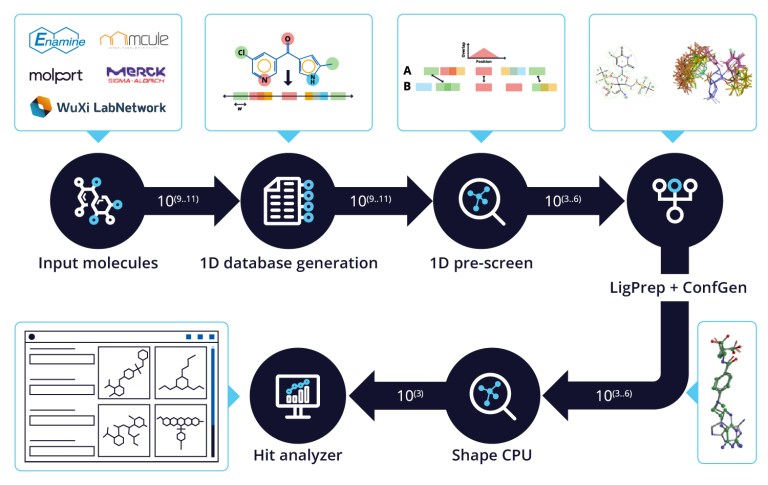 White Paper
Life Science
White Paper
Life Science
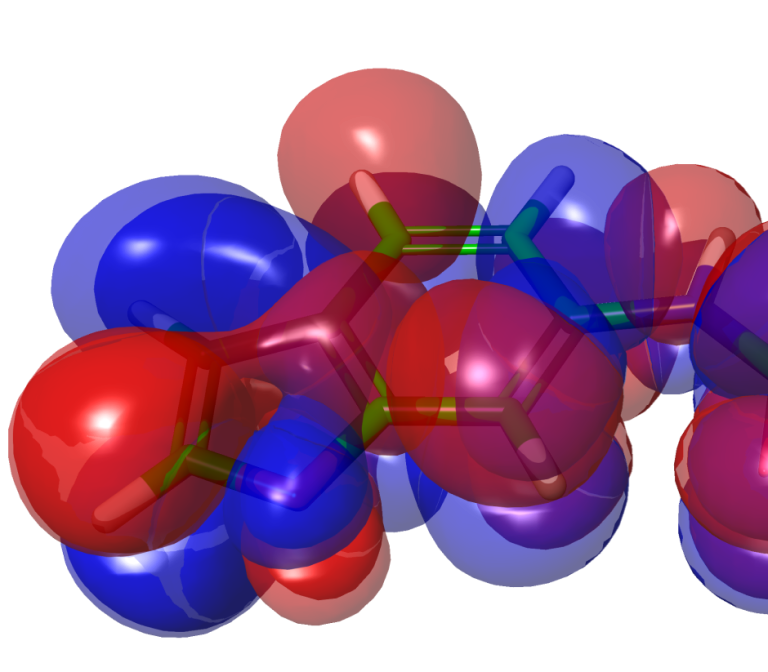 White Paper
Life Science
White Paper
Life Science
Quick Reference Sheets

Latest insights from Extrapolations blog

With FEP+, “The Experiment is the Limit.”
Over the past century, small molecule drugs have represented the dominant modality in drug research, enabling medical breakthroughs that have saved countless lives.

Tackling Drug Solubility: AbbVie and Schrödinger Collaborate to Advance Accurate Prediction Methods
The complexity and size of drug candidates has grown in recent years as scientists pursue novel targets once considered undruggable.
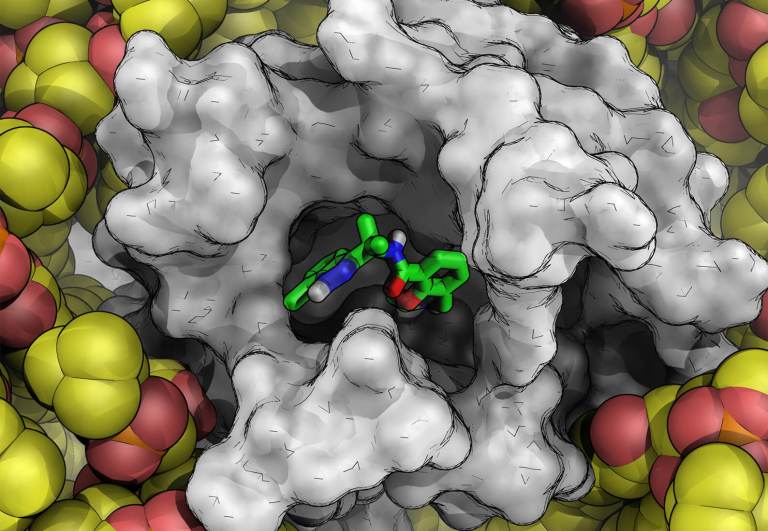 Blog
Blog
Can AlphaFold Models be Used for Structure-Based Drug Design? A Perspective Two Years In
We recently sat down with Edward Miller, Senior Director of Protein Structure Modeling at Schrödinger, to discuss his experience using AlphaFold models for SBDD.
Training & Resources
Online certification courses
Level up your skill set with hands-on, online molecular modeling courses. These self-paced courses cover a range of scientific topics and include access to Schrödinger software and support.
Free learning resources
Learn how to deploy the technology and best practices of Schrödinger software for your project success. Find training resources, tutorials, quick start guides, videos, and more.
Other Resources